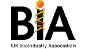NRG Therapeutics is leveraging breakthrough science in mitochondrial biology and disease mechanisms to develop a new approach to tackling neurodegenerative disease by rectifying mitochondrial dysfunction.
In neurodegenerative diseases a key driver of mitochondrial dysfunction and its downstream effects of neuroinflammation and neuronal death is the opening of the mitochondrial permeability transition pore (mPTP) in response to sensitisation or activation by pathogenic proteins such as TDP-43 in ALS/MND and α-synuclein in Parkinson’s.
We are developing disease-modifying mitochondrial therapeutics to slow or halt the progression of neurodegenerative disorders based on inhibition of the mPTP through a novel mechanism of action (MOA) involving an undisclosed mitochondrially-localised protein (“protein A”) that rectifies mitochondrial function.
Discovery: Novel target and MOA
NRG Therapeutics has discovered a novel protein component or regulator of the mPTP.
We have demonstrated that this undisclosed protein (“protein A”) is essential for the mPTP’s opening thereby regulating pore function and mitochondrial integrity.
NRG Therapeutics is focused on developing a pipeline of next-generation mPTP small molecule inhibitors that are orally bioavailable, CNS-penetrant and act by binding to “protein A".
NRG5051 – lead drug candidate
Our lead drug candidate NRG5051 has demonstrated in in vitro assays that it is a potent inhibitor of the mPTP, acting via the novel regulator “protein A”, inhibition of which protects mitochondrial function.
NRG5051 commenced a first-in-human clinical trial in 2026.
Our lead indication is ALS/MND with a planned expansion into additional neurodegenerative diseases such as Parkinson’s.
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by the selective loss of motor neurons resulting in mortality within an average of 2-5 years. Though most cases of ALS are sporadic (sALS), approximately 10% are familial (fALS) in origin.
TDP-43, a DNA/RNA-binding protein that is crucial for normal cellular function, is widely expressed in the brain. Normally it is mostly present in the nucleus but in ALS/MND it incorrectly translocates to the cytoplasm, where it forms mis-folded protein aggregates that are a key pathological feature of ALS/MND.
TDP-43, is the most validated example of a pathogenic protein that activates or sensitises the permeability transition pore and TDP-43 proteinopathy is found in 97% of ALS/MND cases. Blocking this gain-of-function toxicity of mis-localised TDP-43 is therefore expected to have therapeutic benefit in the majority of ALS/MND patients.
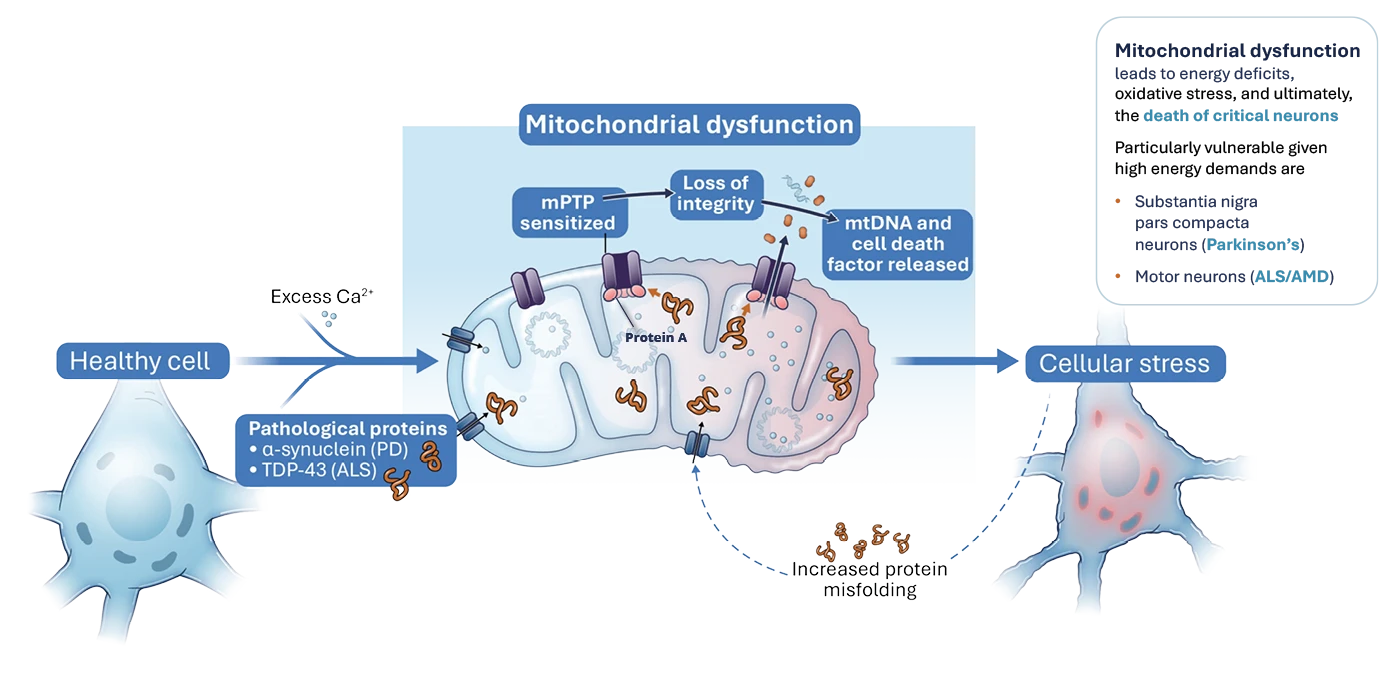
In ALS/MND mitochondrial dysfunction occurs in part as a consequence TDP-43 gain-of-function toxicity, as misfolded TPD-43 induces mPTP opening with two key outcomes:
- mtDNA is released into the cytoplasm and stimulates the cGAS/STING innate immune sensor resulting in interferon signalling and downstream neuroinflammation.1
- The release of cell death factors e.g. cytochrome C is triggered following mPTP opening and collapse of the mitochondria which contributes directly to motor neuron cell death.
NRG5051 has been shown to inhibit cGAS/STING induced innate immune gene expression, improve motor function and reduce neuronal cell death in preclinical models of ALS/MND.
We have generated compelling preclinical data, including a profound effect on the translatable biomarker neurofilament light (NfL) chain, to support NRG5051’s advancement into the clinic for first-in-human trials and as a potential treatment for ALS/MND.
Parkinson’s is one of the fastest growing neurodegenerative conditions with a global prevalence predicted to double by 2050. There are currently no treatments available to slow or halt progression of Parkinson’s, with existing medicines providing temporary symptomatic relief only.
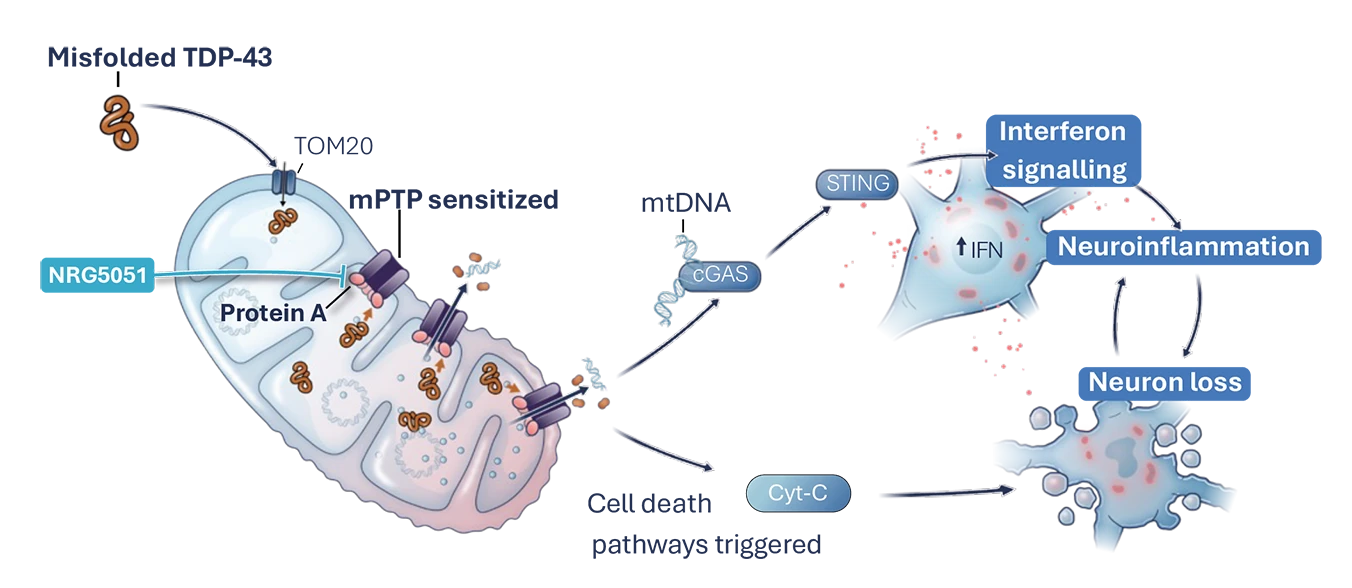
In Parkinson’s, both inherited gene mutations and the toxic pathological protein ⍺-synuclein, impact Ca2+ homeostasis leading to mitochondrial dysfunction and mPTP opening in substantia nigra neurons. As a result, there is a bioenergetic deficit, elevated oxidative stress and neuroinflammation, which ultimately leads to neuronal cell death and disease progression.2,3,4
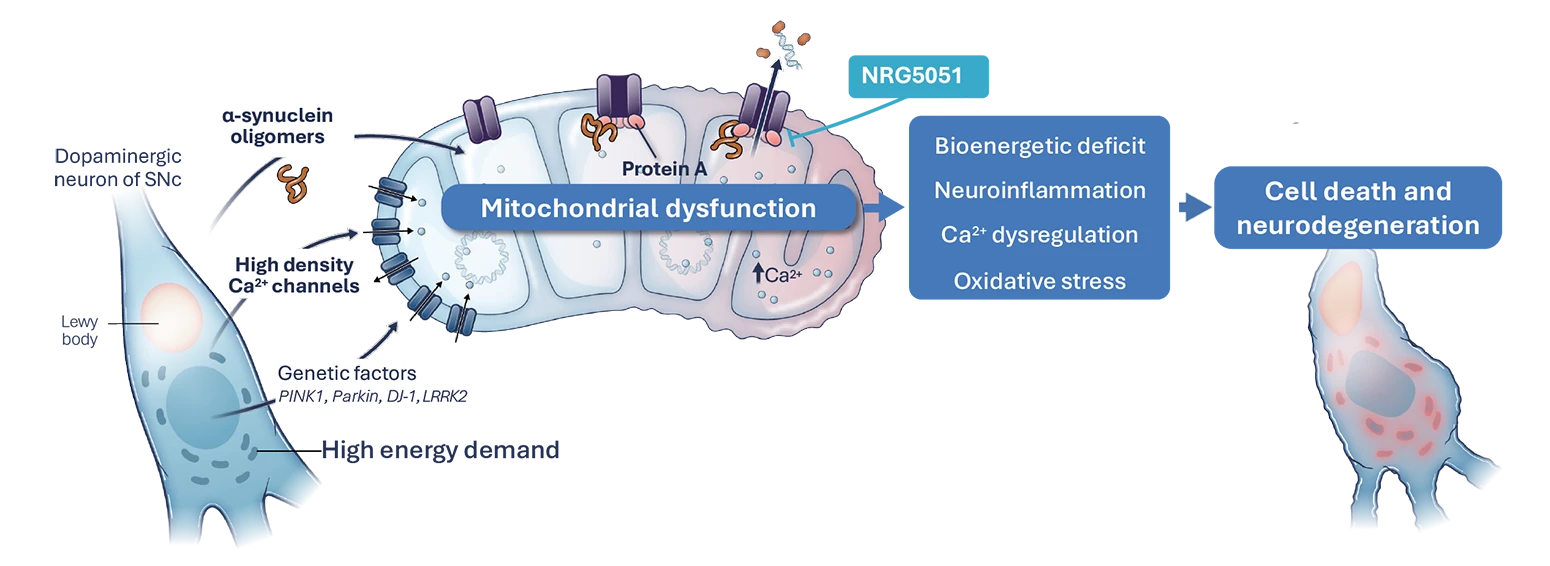
NRG5051 has been shown to inhibit ⍺-synuclein oligomer induced mPTP opening, prevent aspects of neuroinflammation, and is neuroprotective and improves motor function in preclinical models of Parkinson’s.
We have generated compelling preclinical data to support NRG5051’s advancement into the clinic for first-in-human trials and as a potential treatment for Parkinson’s.
1. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS: Yu et al., 2020, Cell 183, 636–649
2. α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease: Ludtmann et al., 2018, Nature Communications, 9, 1-16
3. The Origins of Oxidant Stress in Parkinson’s Disease and Therapeutic Strategies. Surmeier et al., 2011, Antioxidants & Redox Signaling 14, 289-1301.
4. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant α-synuclein transgenic mice. Martin et al., 2014, Neurobiology of Aging 35,1132-52